
DEFINICIÓN
El síndrome de West(SW) o “síndrome de espasmos infantiles” es una encefalopatía (alteración cerebral) de tipo epiléptico que afecta a los niños. Los espasmos infantiles son un tipo especial de ataque epiléptico que inicia en niños menores de un año de edad.
Es una enfermedad poco frecuente, pero severa, que se presenta con mayor probabilidad en bebés prematuros o que tuvieron mala oxigenación cerebral al momento de nacer (hipoxia neonatal), ya que son propensos a sufrir daño neuronal.
Se caracteriza típicamente por tres hallazgos:
- espasmos epilépticos,
- retraso del desarrollo psicomotor y
- electroencefalograma con un trazo característico de “hipsarritmia”.
Dicho padecimiento fue descrito por primera vez en 1841 por el médico británico William James West, quien encontró que su hijo presentaba una forma peculiar de crisis epiléptica (espasmos) y solicitó ayuda a sus colegas a través de un artículo publicado en “The Lancet” (revista médica británica). Desde entonces se han registrado adelantos notables, hay más información y ahora se sabe que se trata de una forma grave de epilepsia con secuelas considerables y de difícil manejo.
Se considera al síndrome de West como un subgrupo dentro del síndrome de espasmos infantiles, que siempre genera algún grado de retraso global en el desarrollo infantil.
¿Qué es la epilepsia?
La epilepsia es un grupo de desórdenes neurológicos caracterizados por descargas eléctricas anormales en el cerebro. Es una enfermedad crónica del sistema nervioso central.
¿Qué es una crisis epiléptica? Es una manifestación clínica producida por la descarga eléctrica excesiva de un grupo de neuronas hiperexcitables, que la mayoría de las veces se presenta como un movimiento del cuerpo brusco y sorpresivo.

En algunos casos conllevapérdida de conciencia, confusión, alucinaciones visuales y auditivas, espasmos, alteraciones sensoriales y en el sistema nervioso automático.
En algunos casos los ataques son precedidos por un “aura”o un sentimiento de inquietud o incomodidad; el aura es parte de la crisis convulsiva y marca el principio del evento en el cerebro. Se conocen tipos diferentes de epilepsia. Para hablar de epilepsia tienen que haberse presentado, cuando menos, dos crisis.
Existen otros tipos de crisis que no se consideran epilépticos como los síncopes nerviosos, las pseudocrisis, los terrores nocturnos, etc.
¿Qué es el retraso psicomotor? Las facultades mentales y la capacidad de movimiento suelen afectarse de forma notable, al grado de que muchos de estos chicos evolucionan nula o lentamente.
¿Qué es el electroencefalograma? El electroencefalograma es un estudio no doloroso y no agresivo en el que se puede registrar y grabar (en un papel especial) la actividad eléctrica del cerebro u “ondas cerebrales”. Las ondas cerebrales es la forma en la que las células cerebrales se “comunican” una a la otra y llevan información del cerebro al resto del cuerpo.
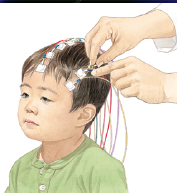
Los electrodos que se le colocan al niño en el cuero cabelludo, registran las ondas eléctricas durante los periodos de actividad y lo ideal sería durante los periodos de sueño, se transmiten al papel donde quedan registrados estos datos (trazado electroencefalográfico).
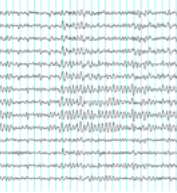
¿Qué es la actividad eléctrica anormal del cerebro tipo hipsarritmia?Cuando ésta se analiza mediante electroencefalograma, se descubre un patrón irregular muy especial, llamado hipsarritmia, que fue descrito desde 1952.
CAUSA DE LA EPILEPSIA
El síndrome de West no es una condición patológica sino que más bien es un síntoma de diferentes desórdenes del cerebro.
Cualquier problema que origine un daño cerebral, puede ser una causa subyacente que desencadene un Síndrome de West y, de acuerdo a esto, las causas pueden ser:prenatales (las más frecuentes), perinatales o postnatales.
CAUSAS PRENATALES:
- La más frecuente (30%) es la displasia cerebral (anormalidad en el aspecto de las células del cerebro). Dentro de esta categoría se incluyen:
- La Esclerosis tuberosa: Es el padecimiento más común responsable del SW. El nombre de esclerosis tuberosa se debe a la formación de tumores no cancerosos en el cerebro, en ojos, en corazón y riñones que tienen forma de raíz y se van calcificando con la edad hasta endurecerse. Otra de las manifestaciones es la presencia de manchas blancas (hipocrómicas) en la piel.
- La Neurofibromatosis: Son trastornos genéticos del sistema nervioso que afectan principalmente al desarrollo y crecimiento de los tejidos de las células nerviosas. Estos trastornos ocasionan tumores que crecen en los nervios.
- El Sindrome de Sturge-Weber: Es un síndrome no hereditario caracterizado por una marca de nacimiento (usualmente en la cara) conocida como mancha en vino de Oporto,y por problemas neurológicos. Entre otras particularidades puede haber, crisis epilépticas.
- La microcefalia congénita: Trastorno neurológico en el cual la circunferencia de la cabeza es más pequeña que el promedio para la edad y el sexo del niño. Entre otros síntomas puede presentar convulsiones.
- El Síndrome de Aicardi: Es un trastorno genético raro caracterizado por contracturas musculares involuntarias, retraso mental, daño oftálmico, además de ausencia (agenesia) del cuerpo calloso del cerebro. Se diagnostica desde los primeros meses de vida.
- La Holoprosencefalia: constituye un amplio espectro de malformaciones del cráneo y la cara debidas a una anormalidad compleja del desarrollo del cerebro. Pueden ocurrir convulsiones o retraso mental.
- La Esquizencefalia: trastorno raro del cerebro, caracterizado por surcos o hendiduras anormales en las dos partes que constituyen el cerebro (hemisferios cerebrales). La mayoría de las personas afectadas sufre convulsiones y algunos pueden presentar hidrocefalia.
- Algunos trastornos cromosómicos (los cromosomas son los pequeños cuerpos en forma de bastoncitos que se encuentran en el núcleo de cada una de las células de nuestro organismo y que llevan toda nuestra información genética). Entre ellos podemos mencionar:
- El Síndrome de Down es un trastorno genético causado por la presencia de una copia extra del cromosoma 21 (o una parte del mismo), en vez de los dos habituales -de ahí el nombre de “trisomía del par 21”- caracterizado por la presencia de un grado variable de retraso mental y unos rasgos físicos peculiares que le dan un aspecto reconocible.
- El Síndrome de Miller Dieker: es una enfermedad rara, congénita y hereditaria. Los afectados presentan problemas en el desarrollo del sistema nervioso central que conduce a alteraciones severas en la función neurológica.
- La duplicación del brazo corto del cromosoma 18 o la del 15.
- Las Infecciones como el citomegalovirus, herpes simple, rubéola, sífilis o toxoplasmosis, cuando afectan al bebé durante el embarazo, pueden ser causa de SW.
- Los Trastornos Metabólicos El metabolismo es el conjunto de reacciones y procesos físico-químicos que ocurren en una célula y en el organismo, para procesar los nutrientes y sacar los deshechos.
- La fenilcetonuria:también conocida como PKU, es un error congénito del metabolismo causado por la carencia de la enzima fenilalanina hidroxilasa, lo que se traduce en la incapacidad de metabolizar el aminoácido tirosina a partir de fenilalanina en el hígado. A los seis meses se hace patente el retraso mental; la mayor parte de los niños afectados son deficientes graves o profundos y en ocasiones se alcanza la deficiencia media.
- Hiperglucemia no ketócica:(HGNC) es un error innato del metabolismo (EIM) que afecta la degradación del aminoácido glicina, el cual se acumula en grandes cantidades en la sangre. Los bebés afectados son normales al nacer, pero en las primeras 24 horas de vida, inician con letargia y/o convulsiones.
- Síndrome de Leigh:es un desorden neurodegenerativo progresivo de inicio temprano con un daño al sistema nervioso característico. Los síntomas dependen de cuáles son las áreas del sistema nervioso central que se encuentran afectadas, puede haber convulsiones.
- Enfermedad de Krabbe:es una enfermedad metabólica que se manifiesta por la aparición de trastornos neurológicos.
- Adrenoleucodistrofia neonatal:esta afección ocasiona la acumulación de ácidos grasos en el sistema nervioso, en las glándulas suprarrenales y en los testículos, lo cual interrumpe la actividad normal y produce cambios en el tono muscular, con espasmos y debilidad progresiva.
- Síndromes congénitos:
- Enfermedad de Fahr: también conocida como ferrocalcinosis cerebro vascular o calcinosis de los núcleos del cerebro.Es una enfermedad que se presenta por calcificaciones masivas de núcleos de sustancia gris central, sin haber anomalía alguna en el metabolismo del calcio. Paralelamente, aparecen signos neurodegenerativos.
- La hipoxia (falta de oxigenación del cerebro en el momento de nacer) o la isquemia (disminución transitoria o permanente del riego sanguíneo en el cerebro) de causa prenatal, en ocasiones son causantes de la aparición del síndrome.
CAUSAS PERINATALES
Se definen como causas perinatales aquellas que tienen lugar entre la semana 28 del embarazo y la primera semana de vida tras el nacimiento. Se incluyen aquí:
- necrosis neural: Una de las formas de muerte neuronal. Esto puede suceder por falta de oxigenación (anoxia) o por tóxicos. La neurona muerta termina destruyéndose y esto causa inflamación.
- status marmoratus: Es una alteración observada en recién nacidos que presentaron trauma en el nacimiento o por inflamación del sistema nervioso central, específicamente en la región conocida como: los ganglios basales. La mayoría de los bebés que la presentan desarrollarán en el futuro alguna forma de parálisis cerebral.
- leucomalacia periventricular: Es un tipo de lesión cerebral que involucra la muerte de pequeñas áreas de tejido cerebral. Es mucho más común observarlo en bebés prematuros que en recién nacidos a término.
- porencefalia: Es un trastorno del sistema nervioso central que involucra un quiste o una cavidad en un hemisferio cerebral en la mayoría de los casos producido por un infarto durante los primeros meses del embarazo.
CAUSAS POSTNATALES DE LA EPILEPSIA
- Infecciones: meningitis bacteriana (inflamación de las membranas que recubren el cerebro), absceso cerebral, meningoencefalitis por virus (sarampión, varicela, herpes simple, enterovirus -lapoliomielitis era la enfermedad más significativa causada por un enterovirus-, adenovirus –(causantes de enfermedades respiratorias) y citomegalovirus.
- Hemorragias o traumatismos con consecuencia de hemorragia subdural (derrame de sangre de los vasos sanguíneos de la duramadre, la más externa de las tres capas –meninges- que cubren al cerebro) o subaracnoidea (derrame de sangre en el espacio subaracnoideo donde normalmente circula líquido cefaloraquídeo).
CLASIFICACIÓN DEL SW
El grupo de trabajo para la Clasificación y Terminología de la Liga Internacional contra la Epilepsia (ILAE) clasifica al SW, según su etiología o causa, en:
- Sintomático: Se denomina SW sintomático al cuadro debido a una o varias lesiones estructurales del cerebro que pueden identificarse. Es el más frecuente, ya que en la actualidad es posible encontrar, en muchos casos, la lesión estructural causante del cuadro.
- Idiopático: En este grupo se encuentran entre el 5 y el 10% de los pacientes con SW en los que no se identifica etiología y no parecen padecer una encefalopatía oculta. Estos niños no tienen antecedentes prenatales o perinatales y su desarrollo psicomotor es normal hasta el comienzo de los espasmos. Se caracterizan por presentar espasmos e hipsarritmia simétrica. En general, el deterioro psicomotor es leve.
- Criptogénico: Se reserva el término “SW criptogénico” para los casos en los que el médico no encuentra evidencia de una alteración cerebral, pero, por la evolución del niño, parece haber una afectación cerebral ‘oculta’ no identificada. En general, estos niños tienen un retraso en el desarrollo psicomotor previo al comienzo de los espasmos.
Otra clasificación muy empleada es la que habla de:
- síndrome de West primario(el que aparece antes de los 3 primeros meses de vida).
- secundario(a partir de los 7-8 meses) y
- tardío(a partir de los dos años).
En el 90% de los casos, las crisis aparecen antes del primer año de edad y en un 10%, antes del tercer mes. Lo más frecuente es entre los 4 y 7 meses de edad, con una mayor frecuencia en el 6° mes. Los inicios más tardíos (después de los 18 meses), generalmente son errores de diagnóstico. Un inicio muy precoz (antes del 3er mes de vida), puede verse en las formas sintomáticas.
SÍNTOMAS
Las crisis epilépticas del Síndrome de West han sido denominadas de diversas formas: espasmo de flexión, mioclonía masiva en flexión, mioclonos infantil, crisis de Salaam y otros.
Los síntomas típicos son:
- Espasmos musculares involuntarios que ocurren por episodios de descargas eléctricas incontrolables en el cerebro (crisis epiléptica).
- Pueden ser espasmos de flexión, de extensión y mixtos.La contracción más típica es la de flexión. Cada movimiento involuntario puede presentarse al despertar o después de alimentar al bebé. Inicia súbitamente ygeneralmente son bilaterales y simétricos de los músculos del cuello, tronco y extremidades. Estos ataques generalmente se presentan en racimo o seguidilla, uno después del otro con un interval de 10 a 15 segundos por varios minutos (puede alargarse a 10-20 minutos). Estos episodios pueden presentarse varias veces al día y es muy común que, al iniciar los espasmos, el niño tenga un cambio notorio en su comportamiento y deje de hablar y de hacer contacto visual.La duración, intensidad y los músculos afectados por los espasmos, varían en cada niño.
- Los espasmos pueden acompañarse de:
- Alteraciones respiratorias.
- Gritos
- Rubor
- Movimientos oculares.
- Sonrisa
- Muecas
- Retraso psicomotor: incluye el retraso para adquirir aquellas capacidades que requieren coordinación de los músculos y movimientos voluntarios y pérdida de habilidades adquiridas y, anormalidades neurológicas como
- Diplejia (parálisis que afecta a partes iguales a cada lado del cuerpo).
- Cuadriplejia (parálisis de los cuatro miembros: tetraplejia).
- Hemiparesia (debilitamiento o ligera parálisis de una mitad del cuerpo).
- Microencefalia (cabeza pequeña).
- Electroencefalograma característico: Como ya lo mencionamos con anterioridad, los niños con Síndrome de West muestran en el electroencefalograma un patrón de ondas cerebrales muy particular llamado hipsarritmiascaracterizado por enlentecimiento y desorganización intensos de la actividad eléctrica cerebral.
Estos tres elementos aparecen a lo largo de varias semanas en un niño hasta entonces normal, o bien que ya ha presentado crisis o signos neurológicos deficitarios (SW secundario).
En un 85% de los casos, inicia con los espasmos y en ocasiones, con la detención del desarrollo psicomotor. El trazado hipsarrítmico del EEG puede faltar al principio o bien descubrirse una vez que la aparición de los espasmos está muy avanzada. En los casos típicos, el síndrome en conjunto se completa en 4 a 6 semanas.
En el curso del proceso, los lactantes pierden la sonrisa, abandonan la prensión de los objetos y el seguimiento ocular. Se vuelven irritables, lloran sin motivo y duermen peor. Disminuye el tono muscular y si la situación se prolonga, el deterioro es importante.
El síndrome -con o sin tratamiento- en un 30% de los casos evoluciona a la forma de epilepsia de Lennox-Gastaut, que se caracteriza por tener hasta tres diferentes tipos de crisis y son difíciles de controlar. Es por esto que es sumamente importante el diagnóstico temprano.
Es frecuente que en muchos niños el retraso mental se manifieste antes que los espasmos; estos casos, en general, se consideran como probablemente sintomáticos.
TIPOS DE CRISIS EPILEPTICAS
La expresión de los espasmos depende de la musculatura afectada y de la duración de cada espasmo. La utilización del video ha permitido un análisis más detallado y riguroso.
Crisis típicas: gran parte de los autores admiten que en la mayoría de los casos (del 68 al 80%), los espasmos se realizan en flexión. Pueden ser más o menos extensos y afectan la musculatura del cuello, del tronco y de los miembros; el niño “se dobla como una navaja al cerrarse”. Flexión de la cabeza y del tronco, los miembros superiores se entrecruzan sobre el pecho, los miembros inferiores en triple flexión.
Si el espasmo se realiza en extensión, el cuello y el tronco se extienden y los miembros se colocan en extensión-abduccion separándose del tronco en forma de cruz.

Sin embargo, lo más frecuente es que sean mixtas –en flexión-extensiónaparentando entonces una “mioclonía masiva” (contracciones breves y bruscas (en relámpago) de un músculo o un grupo de músculos).
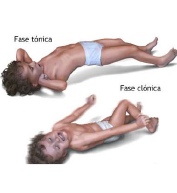
Espasmos de Salaam: Son contracciones en las que el niño parece que va a dar un abrazo (posición de salaam), a la vez que eleva las piernas sobre la pelvis, desvía sus ojos hacia arriba y realiza alguna mueca. Esto puede ocurrir en repetidas ocasiones (en racimo), llegando a sumar hasta 100 en unos minutos.
Crisis atípicas: a menudo limitadas o frustradas en su expresión, corren el riesgo de ser ignoradas o malinterpretadas: sacudidas de cabeza, breve contracción de la musculatura abdominal, elevación de los hombros, simple movimiento de aducción o abducción de los brazos; más raramente el espasmo puede ser asimétrico. Excepcionalmente, la crisis puede expresarse por una disminución notable de los movimientos espontáneos y lentitud extrema de aquellos que son voluntarios (akinesia) y una disminución de la reactividad.
En general, un mismo niño presenta varios tipos de espasmos. Ciertos fenómenos pueden acompañar a las crisis: desviación ocular, alteración respiratoria, alteración del ritmo cardiaco. Un grito o una sonrisa inmotivada, concluyen las crisis.
Debido a la brevedad del espasmo, es difícil evaluar el nivel de conciencia; en las crisis más largas o atípicas, se admite una obnubilación pasajera.
Si en el inicio de la afección los espasmos pueden ser aislados, esporádicos y de aparición preferente al despertar o durante el sueño, en la fase de estado son características las salvas o racimos. En el curso de una determinada salva, la intensidad y la frecuencia aumentan progresivamente para, a continuación, disminuir hasta desaparecer.
Se consideran «gatillos» o desencadenantes el hambre, la excitación, una temperatura elevada o estímulos táctiles o sonoros.
REGRESIÓN PSICOMOTORA
Constituye la segunda manifestación característica del Síndrome. Inicia por una modificación de la conducta.
En varios días, el niño presenta una indiferencia total: pérdida de la sonrisa, de las reacciones a los estímulos sensoriales. Frecuentemente el niño es calificado de sordo o ciego con pérdida de la mímica e inmovilidad motora absoluta.
En el niño de más edad, el desinterés se extiende a los objetos que le son presentados. Este desinterés, esta falta de actividad, dan lugar en ocasiones a actividades estereotipadas; más raramente se observan reacciones de irritabilidad o de agresividad.
Los padres confirman el deterioro, el niño no progresa más y no presenta ninguna nueva adquisición. Por el contrario, pierde las actividades que poseía al inicio de su enfermedad: ya no se sienta, no sostiene la cabeza, no sonríe más, etc.
La pérdida de los contactos más elementales y la propia atonía (pérdida del tono muscular) generalizada, son estigmas de una regresión evidente si aparecen en un niño cuyo desarrollo psicomotor era ya inquietante.
ELECTROENCEFALOGRAMA (EEG)
Las alteraciones del EEG (hipsarritmia) constituyen el tercer criterio fundamental para el diagnóstico del SW, pero NO son específicas de este síndrome. Sin embargo, es un dato constante, si se repiten los EEG, que existe en un momento u otro de la evolución del SW.

Los hallazgos electroencefalográficos más específicos del SW son el enlentecimiento y la desorganización de la actividad eléctrica cerebral, en forma de trazado caótico con mezcla de puntas y ondas lentas independientes. A este patrón característico se le denomina hipsarritmia.
¿Cuántos tipos de hipsarritmia hay?
Hipsarritmia típica: el trazado de la hipsarritmia típica cuando el niño está despierto (estado de vigilia), “está constituido por una sucesión ininterrumpida de ondas lentas y de puntas de gran amplitud, asociadas proporcionalmente, sin relación de fase y distribuidas por todo el cuero cabelludo sin ninguna sincronía” (Gastaut 1964).
Se trata entonces de un trazado totalmente anárquico, caótico, sin regularidad cronológica, morfológica o topográfica, sin el ritmo de base normal de la edad, que llama la atención por su continuidad y que NO asume nunca un carácter paroxístico propiamente dicho.
El carácter bilateral pero asincrónico, con anomalías que pueden pasar de un hemisferio cerebral al otro, de una región a otra, es decir, el “carácter de focalización múltiple y móvil”, había sido considerado por Gastaut y Roger como la manifestación específica de la hipsarritmia (es importante insistir en que la hipsarritmia en sí No define al SW).
El trazado se modifica constantemente en el curso del sueño: tendencia a una organización de puntas-onda rítmica y bisincrónica con fragmentación de la hipsarritmia.
De esta manera, el sueño atenúa la hipsarritmia y no posee interés diagnóstico cuando el trazado en el estado de vigilia es típico. Por el contrario, el trazado en el sueño adquiere un gran interés cuando existen espasmos y un EEG no significativo en el momento en el que el bebé está despierto, o bien cuando en el curso de la evolución, los trazados de vigilia se normalizan.
Hipsarritmia atípica: la hipsarritmia puede ser atípica (30% de los casos), y presentar un trazado excepcionalmente fragmentado en el estado de vigilia, muy lento, con pocos elementos puntiagudos (disrritmia lenta mayor), o por el contrario, muy rápida. Igualmente puede presentar una amplitud asimétrica o asociada a anomalías focales.
La hipsarritmia puede desaparecer y esto explica el interés del médico especialista por repetir los trazados con intervalos de varios días y, sobre todo, de repetir los trazados durante el sueño.
En los síndromes de West con inicio precoz, la hipsarritmia es menos frecuente y menos característica.
Por último, se pueden encontrar aspectoshipsarrítmicosen personas que tienen epilepsia pero que no tienen el Síndrome de West y en personas que tienen encefalopatías (cualquier proceso o trastorno patológico del cerebro) sin epilepsia.
EEG: excepcionalmente el trazado no muestra ninguna variación en el curso del espasmo. No hay una correlación estricta entre un tipo de espasmo y una alteración del trazado.
DIAGNÓSTICO DE LA EPILEPSIA
En la actualidad no se hace el diagnóstico prenatal.
Ante todo, lo importante es que, en cuanto los padres descubran que un niño tiene problemas neurológicos o de desarrollo y que éstos se acompañan con algún tipo de convulsiones, busquen la ayuda de un neuropediatra (médico pediatra especialista en neurología), es fundamental que se establezca un diagnóstico precoz para poder iniciar el tratamiento cuanto antes, pues al desaparecer los espasmos y la hipsarritmia, se produce simultáneamente una mejoría en la actividad psicomotora
Para elaborar su diagnóstico, el médico tiene una importante pista en la presencia de la triada de síntomas característica de este Síndrome que se mencionaron anteriormente.
Es importante que establezca un diagnóstico diferencial con algunos padecimientos no epilépticos como:
- Cólicos del lactante: se refiere a los cólicos con dolor por las contracciones espasmódicas del intestino que van acompañadas de llanto.
- Mioclonía (contracciones breves y bruscas del músculo que van seguidas de relajación) benigna de la infancia temprana.
- Postura de contracción por espasticidad (es un síntoma que refleja un trastorno motor del sistema nervioso en el que algunos músculos se mantienen permanentemente contraídos. Dicha contracción provoca la rigidez y acortamiento de los músculos e interfiere sus distintos movimientos y funciones).
- Reflujo gastroesofágico (es una afección en la cual los contenidos estomacales se devuelven desde el estómago hasta el esófago -el conducto que va desde la boca hasta el estómago- después de comer. Provoca dolor intenso y el bebé se contrae).
Igualmente debe hacerlo con algunos padecimientos epilépticos como:
- Epilepsia mioclónica del lactante (se trata de un tipo de epilepsia que se caracteriza por sacudidas bruscas, bilaterales (únicas o repetitivas) de las manos o de los hombros, que ocurren al despertar, durante el aseo o durante el desayuno).
- Hay ocasiones en que esta enfermedad es antecedida por el síndrome de Ohtahara, que ocurre en la etapa neonatal y se caracteriza por crisis con espasmos, severo atraso psicomotor y escasa respuesta a estímulos sensoriales.
ESTUDIOS DE IMAGEN Y DE LABORATORIO
Para hacer el diagnóstico, el neuropediatra debe caracterizar los patrones de la actividad del cerebro a través de la medición por medio de varios estudios de imagen y laboratorio. Entre ellos, además de la Electroencefalografía, podemos considerar:
Ultrasonido craneal: Se utiliza el ultrasonido al igual que en el embarazo. La fontanela del cerebro es como una ventana por la que el especialista puede ver el tamaño de los ventrículos (áreas llenas de líquido dentro del cerebro) y comprobar si ha habido hemorragia.
Tomografía de emisión de protones (PET): Este estudio no es tan común como los anteriores. Los escaneos PET, que involucran un acelerador lineal, es especialmente productiva porque le permite al investigador valorar el metabolismo del cerebro.
Resonancia magnética: Utiliza un campo magnético de gran alcance y se crean una gama de imágenes en varios planos. Proporciona una información muy detallada sobre la estructura del cerebro.
Otras pruebas
La infección como causa de espasmos infantiles puede determinarse con análisis de sangre, orina y líquido cefalorraquídeo.
- Análisis de sangre: algunas de las pruebas que se hacen incluyen: análisis de los cromosomas (cariotipo), pruebas para determinar una infección reciente antes o después del nacimiento, química sanguínea (para medir glucosa y algunos otros componentes especiales) y el tamiz metabólico ampliado para descartar Errores Innatos del Metabolismo (EIM).
- General de orina: para detectar infecciones recientes o antes del nacimiento y anomalías en la química del organismo .
- Líquido cefalorraquídeo: este líquido se analiza buscando evidencias de infección o de anormalidades químicas en su composición.
Para examinar la piel, el médico puede utilizar una lámpara de Wood (luz ultravioleta) que, en un cuarto obscuro, puede mostrar lesiones en la piel que algunas veces aparecen en conjunto con un desarrollo anormal del cerebro y pueden ser una de las causas de los espasmos infantiles.
Seguramente el neuropediatra te pedirá que acudas a consulta con un médico genetista que te hará pruebas moleculares genéticas para determinar si existe alguna mutación genética que pueda ser causa de este padecimiento (la mutación en el gen ARX y STK9 (CDKL5) asociados al Síndrome de West ligado al cromosoma X) y el patrón de herencia.
Usa nuestra sección de “directorio de Genetistas” para localizar a un profesional especializado en genética en tu área.
No todos los niños requieren de todas estas pruebas y estudios, algunos necesitarán incluso otros diferentes. Dado que el SW es una condición que tiene muchas causas diferentes, la elección del tipo y número de estudios que se le tengan que hacer a tu bebé estará determinada por sus características particulares.
TRATAMIENTO DE SINDROME DE WEST
El tratamiento del Síndrome de West se dirige a los síntomas específicos que son aparentes en cada niño. Es probable que sea necesario contar con los esfuerzos coordinados de un equipo de especialistas: pediatras, neuropediatras, neurofisiólogos, especialistas en rehabilitación física, genetista y otros dependiendo de los síntomas. Este equipo elaborará un plan de tratamiento que cambiará de acuerdo a las circunstancias.
El tratamiento del síndrome de West se basa en el uso de medicamentos antiepilépticos para ayudar a reducir o controlar diferentes tipos de convulsiones asociadas al SW que se toman por tiempo indefinido. Si el tratamiento falla, el médico puede cambiar por otros
Terapia física: Se recomienda también que el bebé reciba estimulación temprana y terapia de rehabilitación cuanto antes donde se deberá trabajar la estimulación del tono muscular, reflejos primitivos, desarrollar plan de ejercicios de coordinación, equilibrio y movilidad.
Cabe señalar que los niños siempre quedan con alguna secuela, pero se sabe que hay pequeños con deficiencias en lenguaje, para caminar o de aprendizaje, que han recuperado buena parte de sus funciones.
Un factor extra que se propone para este padecimiento, es el amor que se le debe dar al niño. En efecto, el chico puede tener retraso, mostrarse triste o indiferente, pero percibe a través de la piel, entiende cómo se le trata y se expresa de manera especial con su mirada; sabe cuándo es rechazado y distingue los sentimientos de sus padres hacia él.
Medicamentos: En un principio el médico iniciará con Piridoxina que es una vitamina del complejo B, para descartar la posibilidad de crisis por falta de esavitamina.La B6 es una vitamina hidrosoluble, y desarrolla una función importantísima que es la síntesis de carbohidratos, proteínas, lípidos y la formación de glóbulos rojos, células y hormonas dentro del organismo.
Vigabatrina: Es la primera elección para el tratamiento de la enfermedad porque tiene menos efectos tóxicos que los otros medicamentos y se han asociado menos recaídas.
Siendo un fármaco eficaz, su uso se ha visto frenado por la posibilidad de ocasionar déficit de visión por reducción de los campos visuales. Los niños deben ser sometidos a exámenes oftalmológicos periódicos.
Esteroides:
ACTH: Fue el primero en demostrar su eficacia en el tratamiento de la enfermedad. Las dosis y esquemas de administración varían enormemente. Presenta una tasa de respuesta similar a los corticoides pero debe valorarse su empleo debido a la toxicidad que puede provocar:
- Mortalidad atribuible al tratamiento es aproximadamente del 5% por hemorragias secundarias a hipertensión o infecciones
- Se detectan cada vez con mayor frecuencia anomalías cardiacas y otros efectos secundarios habituales son el hirsutismo, sedación, y somnolencia acompañada de gran irritabilidad
Prednisona: Menos eficaz que la ACTH en el tratamiento de las convulsiones. Se acompaña frecuentemente de efectos secundarios
- Apetito
- Aumento de peso
- Irritabilidad
- Presión arterial alta
- Nivel de azúcar en sangre elevado
- Nivel de potasio en sangre disminuido
Ácido Valpróico: Es un buen controlador de las crisis hasta en un 50% de los casos. Las dosis empleadas varían mucho. Puede tener un efecto preventivo en los trastornos de conducta. Los efectos secundarios, que son comunes, incluyen aumento del apetito y malestar estomacal. Muy ocasionalmente puede haber daño al hígado, por lo que se recomienda realizar pruebas hepáticas cada 6 meses.
Topiramato: fármaco antiepiléptico de amplio espectro que ha demostrado tasas de respuestas altas y buena tolerancia cuando se introduce progresivamente.
Dieta cetógena: Es una dieta muy estricta y a base de grasas, que hace que se eleven los ácidos cetónicos en sangre. Estos ácidos deprimen la función cerebral y de las neuronas y, por lo tanto, ejercen de antiepilépticos.
Se considera que esta dieta es incompatible con el desarrollo sano del niño aunque en algunos casos es beneficiosa.
Como cualquier tratamiento, las dietas especiales pueden tener efectos secundarios potenciales, y existe un riesgo de causar una deficiencia de requisitos dietéticos esenciales
Otros fármacos, como el felbamato, el nitracepam y el clonacepam, no han demostrado gran utilidad.
Cirugía: En los niños que hay una lesión evidente en el cerebro y en los que se comprueba que la lesión es la responsable de la enfermedad, la cirugía puede ser un último esfuerzo para controlar el espasmo.
La cirugía puede también beneficiar a algunos niños con los espasmos infantiles que no responden a otros tratamientos, y donde hay una sola área de anormalidad de desarrollo del cerebro
¿Qué pasa si no se le da tratamiento? Sin tratamiento y medicamentos es muy raro que se controle y, en cambio, se incrementa el riesgo de que el pequeño desarrolle enfermedades respiratorias o hipertensión endocraneal (aumento de la presión al interior de la cabeza que afecta el funcionamiento cerebral), mismas que pueden desencadenar su muerte.
PRONÓSTICO
El pronóstico global del Sindrome de West, es grave. El retardo mental ocurre en el 90% de los casos y con frecuencia se asocia con déficit motor, trastornos de conducta y rasgos autísticos.
Del 55 al 60% evolucionan posteriormente a otro tipo de epilepsia como el síndrome de Lennox-Gastaut y epilepsias con crisis parciales complejas.
Criterios de pronóstico:
- Edad: El inicio de los espasmos antes de los 3 meses, conduce a un retraso mental profundo.
- Causa: todos los investigadores destacan el peor pronóstico de las formas secundarias. Un factor importante que contribuye al pronóstico, es si el niño afectado se clasifica como criptogénico/idiopático o sintomático, el pronóstico es mejor en los primeros. Sin embargo, en los casos criptogénicos, la demora en el inicio del tratamiento, puede asociarse con un peor pronóstico desde el punto de vista cognitivo.
- Tipos de crisis: cuanto más corto sea el periodo de espasmos, mejor será el pronóstico. Son frecuentes las recaídas después del tratamiento.
- El tratamiento: un tratamiento precoz y adecuado evita el riesgo de recaída e influye en el pronóstico a largo plazo.
- El estado de salud anterior del niño.
En las formas secundarias, los niños desgraciadamente tienen mal pronóstico ya que muchos de ellos presentarán crisis no controladas y un retraso mental severo.
Pero aunque el pronóstico no siempre es favorable, con terapias adecuadas y la colaboración de la familia es posible lograr un buen control y condiciones que permitan mejoría del menor.
INCIDENCIA
El Síndrome de West es un desorden neurológico raro que afecta más a los hombres que a las mujeres (2×1). La forma ligada al cromosoma X afecta más a los varones.
Se estima que el Síndrome de West afecta a 2.5 a 6 de cada 10,000 nacimientos y es responsable de, aproximadamente, el 30% de todos los casos de epilepsia que afectan a los infantes. (NORDS).
Es muy raro que este padecimiento se repita en una familia. La transmisión es de tipo autosómico recesivo (los dos padres tienen que pasarle el gen defectuoso al bebé), estando raramente ligado al sexo.
DIFERENCIAS ENTRE LOS SÍNDROMES DE OTAHARA, WEST Y LENNOX-GASTAUT
Hay ocasiones en que el síndrome de West es antecedido por el síndrome de Otahara, que ocurre desde el mes de nacimiento y se caracteriza por crisis con espasmos, severo atraso psicomotor y escasa respuesta a estímulos sensoriales.
Y también es posible que evolucione a otra forma de expresión clínica, llamada síndrome de Lennox-Gastaut, Por suerte, en un niño con West bien controlado y al que se le practica electroencefalograma, se puede detectar este cambio a tiempo para realizar los ajustes pertinentes.
Otahara: Las convulsiones solamente son mioclónicas, el espasmo parece como si el niño quisiera agarrar una pelota con su cuerpo y se flexiona o se estira como si se asustara. A veces se confunden con cólicos del lactante.
Lennox-Gastaut: El Síndrome de Lennox Gestaut es un desorden raro que generalmente se hace aparente durante la infancia o en la niñez. Se caracteriza por frecuentes episodios de alteraciones eléctricas incontrolables en el cerebro (crisis epilépticas) y retraso psicomotor severo. Es difícil controlar los espasmos y en ocasiones es necesario mezclar más de tres medicamentos. Se presentan más regresiones que van agravándose con el tiempo.
Los individuos afectados con este síndrome, pueden experimentar muchos tipos diferentes de ataques epilépticos. El síndrome de Lennox Gastaut puede ser causado por, o estar en asociación con un número variado de otros padecimientos o condiciones.
ORGANIZACIONES DE APOYO
Estas organizaciones se han establecido para los individuos y sus familias para darles información, apoyo y la oportunidad de establecer contacto con otras personas afectadas
Asociación de Niños West en México Reto a la Vida: Su objetivo es prestar información y apoyo a las personas que padecen SW y a sus familias.Localizada en el Distrito Federal. Tel (01-55) 5740 5215. También puede consultar su página (www.galeon.com/sdrmwest) o escribir al correo electrónico sdrmwest@hotmail.com.
APICE: Asociación Andaluza de Epilepsia: es una entidad privada sin ánimo de lucro. Su objetivo es prestar información y apoyo a las personas que padecen epilepsia y a sus familias. http://www.apiceepilepsia.org
Fundación síndrome de West: nace con la intención de ayudar y asesorar a los afectados por este síndrome y otros afines.http://www.sindromedewest.org
En inglés:
West Syndrome Support Group: Ofrece apoyo e información acerca de este padecimiento y sus consecuencias: http://www.wssg.org.uk/
El contenido de esta sección fue revisado por el Dr. Antonio Bravo Oro, Neuropediatra, Certificado por el consejo nacional de especialistas en Neurología Pediátrica. Tel 01 44 48 119475.
REFERENCIAS
- Ápice, Asociación Andaluza de Epilepsia, Síndrome de West o Espasmos Infantiles, https://www.apiceepilepsia.org/sindromes-y-tipos-de-epilepsias/sindrome-de-west-o-espasmos-infantiles/
- Orphanet, Portal de Información de Enfermedades Raras y Medicamentos Huérfanos, Síndrome de West, https://www.orpha.net/consor/cgi-bin/Disease_Search.php?lng=ES&data_id=894&Disease_Disease_Search_diseaseGroup=S-ndrome-de-West&Disease_Disease_Search_diseaseType=Pat&Enfermedad(es)/grupo%20de%20enfermedades=S-ndrome-de-West&title=S%EDndrome%20de%20West&search=Disease_Search_Simple
- GARD, Genetic and Rare Diseases Information Center, Síndrome de West, https://rarediseases.info.nih.gov/espanol/13300/sindrome-de-west
- UFM, Universidad Francisco Marroquín, Síndrome de West, https://medicina.ufm.edu/eponimo/sindrome-de-west/
- Fundación Once, Discapnet, El portal de las personas con discapacidad, Síndrome de West, https://www.discapnet.es/areas-tematicas/salud/recursos/faqs/sindrome-de-west
- NORD, National Organization of Rare Diseases, Rare Diseas Data Base, West Syndrome, https://rarediseases.org/rare-diseases/west-syndrome/
- Medscape, Drugs an Diseases, Infantil Spasm (West Syndrome), https://emedicine.medscape.com/article/1176431-overview
- Epilepsy action, “we exist to improve the lives of everyone affected by epilepsy, West Syndrome (infantile spasms), https://www.epilepsy.org.uk/info/syndromes/west-syndrome-infantile-spasms
- WebMD, Health A to Z, Livingt with West Syndrome,https://www.webmd.com/children/west-syndrome-support#1
- MedicinNet, Disease and Conditions, West Syndrome (Infantil Spasms), Syntoms, Causes, Treatment, Prognosis, and life expectancy, https://www.medicinenet.com/west_syndrome/article.htm