
La tirosinemia es un error innato del metabolismo en donde el cuerpo humano no puede romper el aminoácido tirosina.
INTRODUCCIÓN
La tirosinemia es un error congénito del metabolismo, en donde el cuerpo humano no puede romper (metabolizar) el aminoácido tirosina.
DEFINICIÓN
La Tirosinemia es un error innato del metabolismo o enfermedad metabólica caracterizado por un nivel elevado del aminoácido tirosina (uno de los 20 aminoácidos que forman parte de las proteínas) en la sangre.
CAUSA
La causa de este trastorno es la deficiencia o ausencia de una de las enzimas que se requieren para degradar la tirosina. Al no metabolizarse, la tirosina se acumula en los tejidos y órganos lo que trae como consecuencia, problemas médicos muy serios.

¿QUÉ ES LA TIROSINA?
La tirosina es uno de los 20 aminoácidos que utiliza la célula para sintetizar las proteínas. Se forma a partir de otro aminoácido llamado fenilalanina y también proviene de las proteínas de la dieta como la soya, pollo, pavo, pescado y la mayoría de los productos que contienen proteínas.
- Es un precursor de las hormonas del tiroides, de las catecolaminas ( adrenalina, dopamina, noradrenalina) y de la melanina.
- Las catecolaminas son conocidas por su regulación de los estados de ánimo.
- Se ha observado que en niveles bajos de catecolaminas las personas tienden a sufrir ansiedad y depresión.
- La melanina es el pigmento responsable del color del pelo y de la piel.
METABOLISMO DE LA TIROSINA
En condiciones normales la tirosina se metaboliza mediante una serie de reacciones enzimáticas transformándose finalmente en energía.
¿CÓMO SE HEREDA LA TIROSINEMIA?
Este trastorno tiene un patrón de herencia autosómico recesivo, es decir, se necesita que las dos copias de un gen de cada célula estén mutadas. Estos niños heredaron de cada uno de sus padres sanos un gen defectuoso. Los padres, cada uno con una copia de un gen defectuoso, son “portadores”.
- Los portadores no tienen el padecimiento porque el otro gen de este par, trabaja correctamente.
- El riesgo de transmisión de la enfermedad es del 25 por ciento.
- El riesgo es el mismo con cada embarazo.
¿QUÉ GENES METABOLIZAN LA TIROSINA?
Hay 3 genes FAH, HPD y TAT que codifican para las enzimas que degradan la Tirosina en un proceso de cinco pasos. Esta deficiencia lleva a una acumulación tóxica de Tirosina y de sus metabolitos que puede dañar el hígado, los riñones, el sistema nervioso y otros órganos y tejidos.
- El gen FAH fabrica una enzima–fumarilacetoacetato hidrolasa. Su deficiencia no permite la degradación completa de la Tirosina.
- En vez de convertirse a moléculas no dañinas, se acumula en el hígado y en los riñones.
- El gen HPD fabrica la enzima llamada 4-hidroxifenilpiruvato Dioxigenasa, que se encuentra en el hígado y en menor cantidad en los riñones.
- Su deficiencia hace que el metabolito 4-hidroxifenilpiruvato se convierta en un compuesto que daña las funciones de la célula.
- El gen TAT provee instrucciones para fabricar una enzima llamada tirosina aminotransferasa.
- Su deficiencia hace que la tirosina no se degrade en forma eficiente por lo que se elevan los niveles en sangre y orina.
¿QUÉ OCURRE EN EL CASO DE UN NIÑO/A QUE NACE CON UNA TIROSINEMIA?
El bebé nace sin problemas, ya que hasta el momento del parto es su madre la que se encarga de metabolizar las proteínas y ella lo hace bien, aunque sea portadora de una información errónea. Cuando el niño comienza a alimentarse, las proteínas de la leche se degradan y liberan todos los aminoácidos.
- La tirosina no se degrada bien debido al defecto enzimático y las sustancias tóxicas comienzan a acumularse.
- El hígado y el riñón del niño se intoxican poco a poco.
- Llega un momento en que el niño comienza a manifestar la enfermedad dependiendo del tipo de tirosinemia.
¿CUÁNTOS TIPOS DE TIROSINEMIA HAY?
Existen tres tipos de Tirosinemia. Cada uno tiene síntomas característicos y es causado por la deficiencia de una enzima diferente que provocan una acumulación tóxica de Tirosina y sus derivados que pueden dañar el hígado, los riñones, el sistema nervioso y otros órganos y tejidos.
- Tirosinemia Tipo I -la forma más grave- causada por deficiencia de la enzima fumarilacetoaceto-hidrolasa.
- Tirosinemia Tipo II, causada por la deficiencia de la enzima aminotransferasa- tirosina.
- Tirosinemia Tipo III -poco frecuente- causada por la deficiencia de la enzima dioxigenasa-4 hidroxifenilpiruvato.
TIROSINEMIA TIPO I
Los bebés con este trastorno pueden presentar la forma aguda o crónica de este desorden.
FORMA AGUDA DE LA TIROSINEMIA TIPO I
La forma aguda de Tirosinemia Tipo I está presente en el momento de nacer (congénita) o aparece durante los primeros meses de vida. Esta forma es más común y más severa que la crónica.
SÍNTOMAS
La tirosinemia tipo I se manifiesta durante los primeros meses de vida con deterioro hepático severo o luego del primer año de vida con disfunción hepática y acidosis tubular renal asociada a un mal desarrollo y raquitismo.
- Los niños no tratados pueden presentar crisis neurológicas, algunas no reconocidas, que duran entre uno a siete días con:
- Alteración del sistema sensorial, dolor abdominal, neuropatía periférica e insuficiencia respiratoria severa.
- Los individuos afectados con este tipo de trastorno tienen un riesgo incrementado de padecer cáncer de hígado (hepatocarcinoma).
- Es frecuente que el niño muera antes de los diez años de edad por insuficiencia hepática o carcinoma hepatocelular.
OTROS SÍNTOMAS
- Diarrea y heces con sangre.
- Vómitos.
- Crecimiento del hígado (hepatomegalia) y bazo (esplenomegalia).
- Tendencia a herirse fácilmente y sangrar, principalmente de la nariz.
- Ictericia (color amarillento) de piel y de ojos.
- Letargia y/o irritabilidad.
- En algunos casos el bebé desarrolla un típico olor a col hervida o champiñones podridos.
- En ocasiones tienen una persistente hipoglucemia con niveles elevados de insulina, otros tienen un grado bajo de acidosis.
- Presentación de un rápido deterioro de las funciones hepáticas y renales que progresan a una falla hepática aguda provocando alteraciones en la coagulación de la sangre.
FORMA CRÓNICA DE LA TIROSINEMIA TIPO I
Es más rara y se caracteriza por un inicio gradual y menos severo de los síntomas. Pueden no presentarse hasta los seis meses de edad siendo el retraso en el crecimiento el primer síntoma. Muchos niños con Tirosinemia Tipo I desarrollan problemas renales del tipo del síndrome de Fanconi.
- El síndrome de Fanconi es un trastorno raro que se caracteriza por disfunción renal asociado a raquitismo.
- Está relacionado también con episodios de vómito, deshidratación, debilidad y fiebre.
- Cardiopatías y episodios de afectación de muchos nervios (polineuropatía).
- Estos episodios que se consideran como crisis neurológicas, asociados con:
- dolores en piernas y estómago, aumento del tono muscular, vómitos, obstrucción de intestino, latido cardiaco irregular, hipertensión.
- Algunos de los afectados presentan conductas de automutilación como morderse la lengua o friccionar los dientes durante estos episodios.
- En algunos casos, durante estas crisis neurológicas, incluso puede haber una falla respiratoria.
CÁNCER DE LAS CÉLULAS DEL HÍGADO (HEPATOCELULAR).
Los niños que no son tratados con medicamento y dieta y que han sobrevivido a la falla hepática aguda, tienen un riesgo incrementado de desarrollar y morir por cáncer de las células hepáticas (hepatocarcinoma). Las formas agudas y crónicas de la enfermedad pueden aparecer en una misma familia.
DIAGNÓSTICO
Estudios de laboratorio
- BIoquímico: analítica básica y específica en sangre y orina.
- Enzimático: buscando en sangre la falta o deficiencia de las enzimas.
- Genético: Se puede recurrir directamente, si hay alta sospecha clínica y bioquímica, al estudio genético.
- Eco abdominal /Resonancia Magnética: puede haber nódulos hepáticos y aumento del tamaño renal.
TRATAMIENTO
Requiere un equipo médico especializado en enfermedades metabólicas y un nutriólogo. Una parte importante del tratamiento incluye una dieta baja en proteínas que comprenda cantidades limitadas de alimentos que contengan tirosina y fenilalanina.
DIETA
La dieta se inicia de inmediato y depende de la edad, peso y niveles de tirosina en sangre. Dado que las proteínas son esenciales para el crecimiento y desarrollo, se requieren fórmulas especiales. En recién nacidos y lactantes pequeños puede combinarse lactancia materna con estas fórmulas.
- Algunos casos mejoran tan sólo con la dieta.
- Sin embargo, la progresión hacia la cirrosis, falla hepática y un potencial cáncer hepático son aún posibles.
- Por lo tanto, las personas afectadas deben seguir una dieta muy estricta durante toda su vida.
MEDICAMENTOS
La Nitisona NTBC, es el pilar fundamental del tratamiento. La dosis se ajusta según los resultados de los exámenes de laboratorio y al peso del niño. Es muy importante el inicio del tratamiento precoz para una evolución satisfactoria con mínimo riesgo de desarrollo de hepatocarcinoma.
- Debido a que la Nitisona incrementa los niveles de tirosina, el manejo dietético es inmediato para prevenir la formación de cristales en la córnea.
QUIRÚRGICO
Al progresar la enfermedad, puede haber falla hepática que requiera trasplante de hígado. En algunos casos, esto puede ayudar a mejorar incluso el funcionamiento de los riñones. El trasplante hepático puede ser requerido también para prevenir el potencial desarrollo del carcinoma hepático.
- Si no es tratado, la muerte por insuficiencia hepática suele ocurrir en el primer año de vida, generalmente entre los 3 y 9 meses de edad.
CONSEJO GENÉTICO
Se recomienda Consejo Genético para los afectados y sus familias. Es el proceso por el cual el genetista ofrece información acerca del padecimiento, el tratamiento, la forma de herencia y los riesgos genéticos que puedan tener otros miembros de la familia .
- Orienta acerca de las fuentes de apoyo a las que se puedan acercar.
- Ofrece la posibilidad de, además de los exámenes de sangre para detectar la deficiencia de la enzima, realizar pruebas moleculares genéticas.
- De esta manera podrán tomar decisiones médicas y personales informadas.
Usa nuestra sección de “directorio de Genetistas” para localizar a un profesional especializado en genética en tu área.
TRATAMIENTO DURANTE EL EMBARAZO
Se deben llevar a cabo controles bioquímicos muy estrictos con monitorización intensiva de los niveles de fenilalanina y tirosina desde 3 meses antes de quedarse embarazada y durante todo el embarazo para evitar defectos al nacimiento.
- Aunque no hay evidencia de teratogenicidad con pequeñas dosis de NTBC, no se posee experiencia acerca de la dosis durante el embarazo.
- Se ha documentado embarazo e hijo/a sanos en pacientes con trasplante hepático y Tirosinemia I.
- NTBC en varones no es espermatotóxico.
TIROSINEMIA TIPO II (SINDROME DE RICHNER-HANHART) y TIPO III
DEFINICIÓN
La tirosinemia tipo II, es un raro error congénito del metabolismo de la tirosina caracterizado por hipertirosinemia y manifestaciones óculo- cutáneas y, en algunos casos, retraso mental. Se le conoce también como el Síndrome de Richner-Hanhart).
CAUSA
La Tirosinemia Tipo II es causada por mutaciones del gen TAT. Los elevados niveles de tirosina causados por la deficiencia del TAT resultan en el depósito de cristales de tirosina que lleva a los síntomas óculocutáneos. Se sugiere una correlación entre los niveles de tirosina y el daño del SNC.
SÍNTOMAS
Los síntomas se inician en la primera infancia e incluyen:
- Oculares: Lagrimeo excesivo, sensibilidad anormal a la luz (fotofobia), dolor y enrojecimiento de los ojos.
- Los efectos a largo plazo incluyen también: córnea nublada y lesionada, úlceras corneales y cicatrices que disminuyen la agudeza visual.
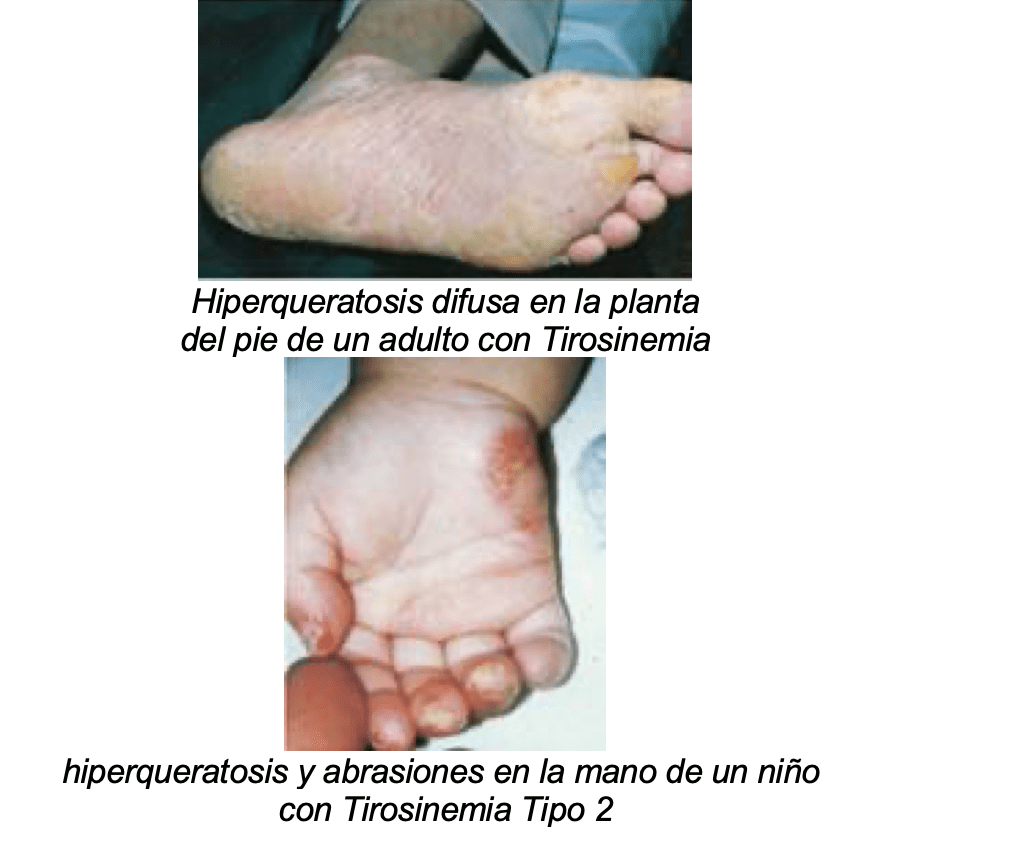
- De piel: generalmente se inician después del primer año pero pueden iniciarse al mismo tiempo que las oculares.
- Pápulas y placas hiperqueratósicas que no dan comezón, dolorosas y progresivas.
- Usualmente están confinadas a palmas y plantas de los pies, especialmente en la punta de los dedos.
- SNC: El retraso mental es variable (de leve a severo). Puede haber problemas de comportamiento, nistagmus, ataxia y convulsiones.
DIAGNÓSTICO
El diagnóstico se basa en los síntomas clínicos y en los exámenes de laboratorio, especialmente las cifras de tirosina y la excreción urinaria de ácidos fenólicos. Se debe confirmar el diagnóstico midiendo la actividad enzimática o analizando las mutaciones genéticas.
EVALUACIONES POSTERIORES AL DIAGNÓSTICO
Para el médico será importante establecer el grado al que se ha extendido la enfermedad por lo que seguramente le hará estudios:
- Análisis de sangre contabilizando plaquetas, electrolitos y funcionamiento del hígado y riñones.
- Tomografía computarizada (TC) de abdomen buscando tumores o nódulos de algún tipo en el hígado y evaluar el tamaño de los riñones.
- RX de muñeca buscando la presencia de raquitismo.
TRATAMIENTO
Dieta con restricción de fenilalanina y tirosina; retinoides orales para el tratamiento de las lesiones cutáneas. La dieta controlada resulta en una rápida resolución de las manifestaciones oculocutáneas. Sin embargo, no queda claro en qué medida la dieta controlada evita la afectación del SNC.
- Como medicamento se puede usar igualmente la Nitizona NTBC para ayudar a controlar la cantidad de Tirosina en el cuerpo.
TIROSINEMIA TIPO III
Es un desorden raro, sólo se sabe de unos pocos casos y se desconoce el espectro clínico así como la causa del retardo mental o los síntomas neurológicos, probablemente sea por los niveles altos de tirosina.
TIROSINEMIA TRANSITORIA DEL NEONATO
10% de los recién nacidos presentan elevados niveles de Tirosina. La causa no es genética. Lo más probable es que haya alguna deficiencia temporal de vitamina C o que, en el caso de un bebé prematuro, las enzimas que produce el hígado inmaduro, no son capaces de realizar su función.
DIAGNÓSTICO
Es importante un diagnóstico precoz que permita iniciar el tratamiento de inmediato y evitar la acumulación de los productos tóxicos. El diagnóstico se realiza en el momento de hacerle el Tamiz Neonatal ampliado cuando se descubre el metabolito succinilacetona en la prueba.
- Es importante que el médico determine el tipo de Tirosinemia que tiene el bebé.
RECOMENDACIONES
Es importante encontrar a un especialista con experiencia en el tratamiento de este padecimiento. Los puedes encontrar en grupos de apoyo, o artículos publicados en revistas médicas. Si tienes alguna duda, envía tu pregunta al Consultorio Virtual donde médicos especialistas te responderán a la brevedad.
REFERENCIAS
- Academic, Diccionario médico, esacademic.com, Tirosinemia Hereditaria, https://esacademic.com/dic.nsf/es_mediclopedia/52688/tirosinemia
- Newbornscreening info, Enfermedades relacionadas con los aminoácidos, Tirosinemia Tipo I, http://www.newbornscreening.info/spanish/parent/Amino_acid/Tyrosinemia.html
- Science Direct, Journals and Books, Anales de Pediatría, Recomendaciones y manejo de la tirosinemia hereditaria Tipo I o Tirosinemia hepatorrenal,https://www.sciencedirect.com/science/article/abs/pii/S1695403310001669
- Anales de Pediatría, Asoiación Española de Pediatría, Recomendaciones y Manejo de la Tirosinemia Tipo I o Tirosinemia hepatorrenal, https://www.analesdepediatria.org/es-recomendaciones-manejo-tirosinemia-hereditaria-tipo-articulo-S1695403310001669
- NCBI Resources, Bookshelf, Tyrosinemia Type I, https://www.ncbi.nlm.nih.gov/books/NBK1515/
- NIH, GARD, Genetic and Rare Diseases Information Center. https://rarediseases.info.nih.gov/diseases/3105/tyrosinemia-type-2